

When a biologic or temperature-sensitive drug loses potency in storage, failure is rarely sudden. It accumulates through uncontrolled freeze–thaw cycles, poorly characterized freezing fronts, and formulation choices made without adequate stress-testing.
For development and manufacturing teams operating under European Medicines Agency and Bundesinstitut für Arzneimittel und Medizinprodukte oversight in Germany, this risk is concrete. Freezing is a regulated manufacturing step, and gaps between process understanding and documented validation surface quickly during GMP inspections and marketing authorization reviews.
Approximately 50% of biopharmaceuticals approved by the U.S. Food and Drug Administration and the EMA are stabilized using lyophilization as a primary method. Separately, over 70% of antibiotics on the World Health Organization’s Essential Medicines List are supplied as sterile lyophilized injectables. These figures reflect how freezing and freeze-drying function as standard, high-impact processes with direct CMC and regulatory consequences.
This blog explains which Pharmaceutical Freezing Solutions apply to different product types and how they must be controlled and documented for EMA and BfArM submissions.
Why Freezing Is a Critical Process Parameter for Drug Stability
Freezing is a manufacturing step with direct consequences for Critical Quality Attributes (CQAs). For proteins, monoclonal antibodies, and complex drug substances, every freeze-thaw (F/T) cycle introduces physical stress that can compromise molecular integrity.
When these stresses are uncontrolled, the downstream consequences include:
- Protein aggregation or unfolding that reduces biological activity.
- Degradation of the active pharmaceutical ingredient (API).
- pH shifts caused by the differential crystallization of buffer components.
- Increased immunogenicity risk in injectable biologics.
- Residual moisture levels above specification in lyophilized products.
- Failed batch release testing and associated documentation gaps.
From a Chemistry, Manufacturing, and Controls (CMC) perspective, freezing parameters must be identified as Critical Process Parameters (CPPs), validated at each scale, and documented in a manner that supports both national BfArM filings and EMA centralized marketing authorization applications (MAAs).
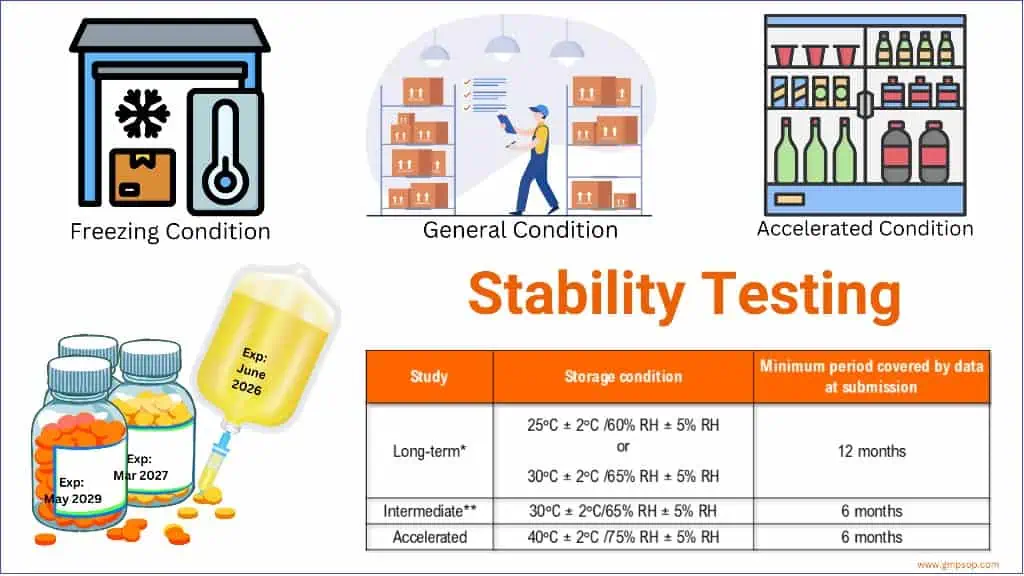
ICH Q1A(R2), the primary guideline governing stability testing, requires that freeze-thaw cycling studies be included as part of the stress-testing package. These studies must demonstrate that the proposed storage conditions are justified by stability data across the full product lifecycle.
Core Pharmaceutical Freezing Technologies and Their Applications
Different drug modalities require different freezing strategies. Selection must account for the product’s thermal stability range, intended storage conditions, and the regulatory submission format it will support.
1. Controlled-Rate Freezing
Controlled-rate freezing uses programmable cooling systems to lower the temperature at a defined, reproducible rate. This approach minimizes ice crystal heterogeneity across the container and reduces cryo-concentration stress on sensitive molecules.
It applies most directly to:
- Bulk drug substances are stored in large-format containers before fill-finish operations
- Biologics with narrow thermal stability profiles.
- Products transferring from laboratory scale to GMP pilot or commercial scale, where process parameters must be re-characterized.
A key variable in controlled-rate freezing is the last point to freeze (LPF), the final area within the container to solidify. Uncharacterized LPF positions create concentration gradients that can affect downstream drying performance and analytical comparability between development and manufacturing batches. This characterization must be included in the process validation package for EMA submissions.
2. Lyophilization (Freeze-Drying)
Lyophilization is the most widely adopted pharmaceutical freezing solution for long-term product stabilization. The process consists of three defined stages, each with distinct CPPs:
| Stage | Description | Key Parameters to Control |
| Freezing | Solidification of the liquid formulation. | Cooling rate, annealing temperature, eutectic point. |
| Primary Drying | Sublimation of ice under reduced pressure. | Shelf temperature, chamber pressure, product temperature. |
| Secondary Drying | Desorption of residual bound moisture. | Ramp rate, end-point moisture specification. |
The EMA’s guideline on lyophilization of parenterals specifies that shelf temperature during primary drying must be maintained below the product’s collapse temperature. Collapse results in moisture entrapment and direct implications for long-term stability.
For products intended for the German and EU markets, the EMA’s decision-tree framework for packaging interaction risk must also be applied. This assesses leachable migration from primary containers during the solid-state and reconstitution phases of the lyophilized product lifecycle.
Post-lyophilization release testing must confirm:
- Residual moisture content within specification.
- Protein conformation integrity.
- Acceptable cake appearance and structure.
- Reconstitution time within labeled limits.
3. Ultra-Low Temperature (ULT) Storage
Certain biologics and investigational medicinal products (IMPs) require storage between -60°C and -80°C. ULT storage is standard for monoclonal antibodies in bulk, clinical-phase biologics, and some advanced therapy medicinal products (ATMPs) in transit.
Cold chain infrastructure for ULT storage must be qualified in accordance with EMA GMP guidelines, specifically Annex 15 on qualification and validation. Temperature mapping, deviation logging, calibration records, and qualification of the continuous monitoring system are all required documentation components.
Under German pharmaceutical law (Arzneimittelgesetz, AMG), any breach in cold chain integrity for authorized products must be assessed for its impact on the risk-benefit profile. This adds a pharmacovigilance (PV) dimension to cold chain management that goes beyond simple operational compliance.
Formulation Design for Freeze-Stable Drug Products
Freezing performance depends as much on formulation composition as on equipment parameters. Excipient selection must be evaluated systematically before moving into GMP-scale manufacturing.
Key formulation variables include:
- Cryoprotectants: Sucrose and trehalose are the most commonly used cryoprotectants in EU-approved biologic formulations. They protect proteins during freezing via vitrification, forming a glassy matrix that limits ice-crystal damage and molecular mobility. Concentration must be optimized as both under- and over-formulation affect structural stability.
- Lyoprotectants: These preserve protein hydrogen bonding during the drying phase when water molecules are removed. Their selection directly determines long-term solid-state stability and must be confirmed through stability-indicating assays.
- Buffer selection and pH management: Certain buffers, including phosphate buffers, crystallize preferentially during freezing, causing pH shifts that can denature proteins. Buffer selection must account for crystallization behavior at sub-zero temperatures. This consideration is particularly important for products targeting parenteral administration, which represent a high-scrutiny category under EMA guidance.
- Surfactants and tonicity modifiers: Polysorbate-based surfactants are frequently included to reduce surface tension and aggregation tendency during freeze-thaw cycles. Their compatibility with primary container materials must be confirmed through extractables and leachables (E&L) studies, a requirement explicitly addressed in EMA’s guideline CPMP/QWP/4359/03 on plastic primary packaging materials.
All excipients must be qualified under ICH Q3C guidelines where applicable, and formulation rationale must be documented in the Quality Target Product Profile (QTPP) and the CMC section of the marketing authorization application.
Regulatory Requirements for Pharmaceutical Freezing in Germany
German pharmaceutical regulation integrates national law under the AMG with EU-level requirements governed by EMA. For manufacturers seeking market access in Germany, both layers apply simultaneously.
BfArM and EMA Submission Requirements
For nationally authorized products, BfArM requires a complete pharmaceutical quality dossier demonstrating that manufacturing processes, including freezing and storage, are controlled, validated, and reproducible. The statutory review timeframe for national marketing authorization applications is seven months upon receipt of a completed dossier.
For products authorized through the EMA centralized procedure, which applies to most biologics and advanced-therapy products, the EMA’s 210-day review process includes a technical assessment of CMC data. Any deficiencies related to process validation, stability, or analytical method validation will generate a List of Outstanding Issues (LoOI) that delays approval timelines.
Post-authorization, any change to a freezing process or storage condition that affects product quality must be submitted as a variation. Depending on its classification, this may be a Type IA notification, a Type IB change, or a Type II variation requiring prior approval. Classification is based on the regulatory impact on safety, quality, and efficacy.
ICH Stability Requirements Applicable to Frozen Products
For frozen drug substances and drug products, ICH Q1A(R2) requirements include:
- Long-term stability testing at the intended frozen storage temperature.
- Accelerated stability studies, where applicable, to assess degradation kinetics.
- Stress-testing, including freeze-thaw cycling studies, to characterize the product’s sensitivity range.
- Photostability testing for transparent primary containers is relevant.
Stability data must be generated using the same container-closure system, fill volume, and orientation as the commercial product. Any deviation from these conditions requires scientific justification within the dossier.
GMP Documentation and Inspection Readiness
Under EU GMP regulations, specifically Annex 1 (manufacture of sterile medicinal products, revised 2022) and Annex 15 (qualification and validation), all pharmaceutical freezing equipment and processes must be qualified and validated before use in GMP. This includes:
- Installation Qualification (IQ) and Operational Qualification (OQ) for all freezing and storage equipment
- Performance Qualification (PQ) runs at maximum and minimum fill loads for lyophilizers
- Temperature mapping of storage environments with documented uniformity data
- Continuous monitoring systems with defined alarm thresholds and deviation management procedures
GMP inspection failures related to cold chain documentation are among the most frequently cited deficiencies in EMA and BfArM manufacturing site inspections. Complete, traceable documentation is not simply a submission requirement. It is an ongoing operational standard.
Analytical Methods for Freezing Process Control and Stability Monitoring
Analytical support for pharmaceutical freezing must be structured, validated, and capable of detecting quality changes resulting from process deviations or stability-indicating degradation.
Standard analytical methods applied across freeze-drying and frozen storage programs include:
| Method | Application | Regulatory Alignment |
| Karl Fischer Titration | Residual moisture measurement in lyophilized products. | ICH Q6A, USP <921>, Ph. Eur. 2.5.12 |
| Differential Scanning Calorimetry (DSC) | Determination of glass transition temperature (Tg) and collapse temperature (Tc). | ICH Q2(R1) |
| Dynamic Light Scattering (DLS) | Aggregation and particle size monitoring in protein formulations. | ICH Q6B |
| Size Exclusion Chromatography (SEC) | Quantification of purity, degradation products, and aggregate content. | ICH Q2(R1), Q6B |
| Circular Dichroism (CD) Spectroscopy | Secondary structure monitoring of proteins after freeze-thaw cycles. | ICH Q6B |
Each method must be validated in accordance with ICH Q2(R1) before use in GMP stability or release testing. Validation data, including specificity, linearity, accuracy, precision, and robustness parameters, must be included in the CMC dossier. For products targeting both German national authorization and EMA centralized approval, method validation must meet the requirements of both the European Pharmacopoeia (Ph. Eur.) and the relevant ICH guideline.
Conclusion
Freezing strategy sits at the intersection of product quality and regulatory accountability. In Germany, it is assessed not only through stability outcomes but through how clearly each decision is justified, validated, and documented.
As expectations from European Medicines Agency and Bundesinstitut für Arzneimittel und Medizinprodukte continue to tighten, manufacturers are evaluated on their ability to demonstrate control across development, scale-up, and routine production. Gaps in freezing rationale or documentation are difficult to defend once a product enters review or inspection.
Teams that embed freezing considerations into development planning rather than treating them as downstream requirements are better positioned to support approval, manage post-authorization changes, and maintain consistent product performance over time.


